Using Rosetta for Ab Initio Structure Prediction in the Fourth Community Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP4)
One of the most important unsolved problems in Molecular Biology is the protein folding problem, which can be stated simply as: given the sequence of amino acids of a protein, what is its three dimensional structure? What makes this problem so important is the fact that the structure of the protein determines its function. However, the gap between the number of known amino acid sequences and the number of known three dimensional structures grows rapidly. One of the reasons for this is the fact that the conventional methods to determine the three-dimensional structures of proteins such as NMR and X-ray crystallography are slow and cumbersome. Therefore a computational solution of this problem is more than desirable. In general, methods that predict the three-dimensional structure of proteins from their sequences are referred to as ab initio protein structure predictions and the problem itself is referred to as ab initio protein folding. Many of the current methods to predict protein tertiary structure use information from the protein database of known sequences and three-dimensional structures of proteins. This information is for example used for the estimation of parameters in knowledge based scoring functions or in the training sets of methods based on machine learning. In our approach, we used knowledge of non-homologous sequences (i.e. sequences that had less than 25% residues in common in pairwise alignments) and their solved three-dimensional structures for the creation of a computer program called ROSETTA, which uses simulated annealing to create protein tertiary structures. One of the key ingredients of ROSETTA is a scoring function that assigns a score to three-dimensional structures and guides the search from the extended chain to a protein-like fold. It captures sequence dependent features of protein structures, such as the burial of hydrophobic residues in the core, as well as universal sequence independent features, such as the assembly of beta-strands into beta-sheets. More details about this scoring function and an evaluation of the folding algorithm can be found in the paper Improved Recognition of Native-like Protein Structures using a Combination of Sequence-dependent and Sequence-independent Features of Proteins (Simons KT, Ruczinski I, Kooperberg C, Fox B, Bystroff C, Baker D. Proteins 34 (1) 82-95, 1999).
For a fair comparison of existing protein structure prediction methods
developed in various labs around the world, a semi-annual community
wide blind test was implemented. For the Community Wide Experiment on
the Critical Assessment of Techniques for Protein Structure Prediction
(CASP), some newly solved three-dimensional structures of proteins are
withheld, and only their sequences are made public. The structure of
those proteins will not be published until a given deadline, which
gives CASP participants the chance to submit their predictions for the
proteins. We (the Baker lab) used our folding routine in this year's
structure prediction competition (CASP4), and it was quite
successful. We produced de novo structure predictions of
unprecedented accuracy, some of which you can check out below.





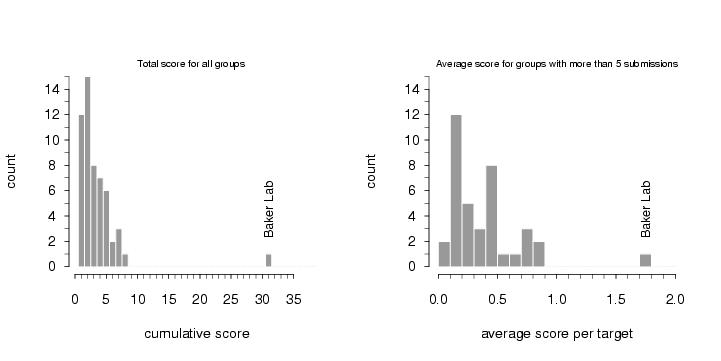
To compare the quality of the predictions of the various groups, the submissions for CASP4 were scored by the organizers (2 points for a largely correct prediction, 1 point for a somewhat correct prediction, 0 points otherwise). The histogram in the left panel below shows the total scores for all groups that submitted predictions for ab initio targets. Since predicting three-dimensional protein structures takes many recources (computer and people time), not all labs were able to submit as many targets as we did, and one could argue that displaying the results as cumulative scores, the way it was done in the histogram on the left, was not completely fair to smaller labs. To account for that, we also show the average score per target submitted (for labs that submitted predictions for more than 5 targets). These data, as a measure of reliability, are shown in the right panel below.